Our research focuses on thoracic aortic aneurysm disease and inherited cardiac arrhythmia
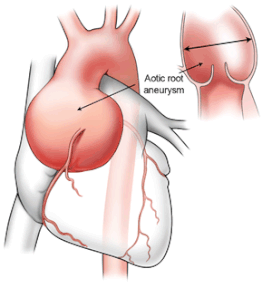
Thoracic aortic aneurysm disease
|
Cardiac arrhythmia
|
Thoracic aortic aneurysm disease
Background
The aorta serves as the responsible artery for blood distribution from the heart towards the distal parts of the human body. A pathological expansion of the thoracic aorta is called a thoracic aortic aneurysm and entails a high risk for aortic dissection and/or rupture. The latter events associate with severe internal bleedings, often resulting in sudden death. To date, genetic defects in more than 30 genes have been linked with thoracic aortic aneurysm, explaining about 30% of patients. Identification and functional characterization of these disease genes have been key in acquiring our current aortopathy knowledge and delivering novel decelerating therapeutic agents. Medical therapies capable of completely stopping or even reversing aneurysm formation are not yet available though.
Goal
In the Loeys lab, we aim to contribute to the further elucidation of the genetic and mechanistic landscape of thoracic aortic aneurysm, with the ultimate goal of improving patient management. The ongoing research lines are contrived in such a way that their results are expected to increase the molecular diagnostic yield, to improve genetic counseling, and to identify predictive markers and curative therapies.
Strategy
The lab has a longstanding and still ongoing tradition in the use of DNA sequencing technologies in affected individuals from families that are negative for mutations in the known genes to find novel thoracic aortic aneurysm genes. For a selection of these genes, we seek to profoundly map the downstream functional consequences and to pinpoint novel drug targets and/or genuine read-outs for drug testing. More recently, a major research line on the discovery and first-line functional characterization of genetic aortopathy modifiers was also established. Individuals belonging to the same family and carrying the same primary mutation can namely range from completely asymptomatic to sudden death at young age due to dissection, considerably complicating patient counselling.
Besides traditional molecular biology approaches, the current projects involve the use of state-of-the-art techniques such as whole exome sequencing, whole genome sequencing, transcriptomics and interactomics/proteomics in patient samples, induced pluripotent stem cell-derived vascular smooth muscle cells and/or mouse models.
Besides traditional molecular biology approaches, the current projects involve the use of state-of-the-art techniques such as whole exome sequencing, whole genome sequencing, transcriptomics and interactomics/proteomics in patient samples, induced pluripotent stem cell-derived vascular smooth muscle cells and/or mouse models.
Cardiac arrhythmia
Background
Inherited cardiomyopathies and primary cardiac arrhythmia disorders are important causes of sudden cardiac death, especially when this is occurring in young individuals. More than 70 genes have already been associated with these disorders, but in more than half of the patients no mutations are detected in any of the known genes and the genetic cause remains elusive. Some insight has been gained in the pathophysiological mechanisms causing inherited cardiomyopathies and primary arrhythmias, but the picture remains far from complete.
Goal
We aim to further investigate the molecular mechanisms underlying cardiac arrhythmia. This will lead to a significantly improved understanding of the disorders, drive the development of novel therapies, improve genetic counseling and result in more accurate risk prediction and personalized management of patients.
Strategy
We will identify novel genes using next-generation sequencing techniques, followed by characterization of their downstream consequences through the use of state-of-the-art cell or animal models. More specifically we study patient-specific induced pluripotent stem cells and transgenic zebrafish or mice. Recently we are also focusing on the identification of genetic modifiers that play a role in the development of the disorders and can explain the phenotypic variability observed within families.